多灶性运动神经病是什么?
多灶性运动神经病(Multifocal motor neuropathy,MMN),又称多灶性脱髓鞘性运动神经病。MMN 作为罕见的纯运动性自身免疫性周围神经病(2018 年列入我国首批罕见病目录),以多灶性运动传导阻滞(CB)为病理核心,全球患病率 0.3-3/10 万,男性高发(男女比 2.7:1),中年(40-50 岁)起病占 70%,儿童罕见。区别于 CIDP(感觉运动均受累)和 ALS(不可逆神经元死亡),其病程呈阶梯式进展或长期稳定,早期干预可显著延缓致残。
多灶性运动神经病的病理机制
1.免疫攻击靶点:30%-60% 患者血清 IgM 型抗 GM1 抗体阳性,靶向郎飞结 / 结旁区 GM1 糖脂,干扰钠 / 钾通道簇集(如 Nav1.6 通道),引发可逆性传导阻滞。最新研究发现,补体 C5b-9 膜攻击复合物沉积是轴突损伤的关键环节。
2.选择性运动神经受累:运动神经 GM1 含量是感觉神经的 3 倍,且结旁区糖脂结构更易暴露抗体表位,解释了纯运动损害的特性。约 10% 抗体阴性患者存在抗 GD1a 等非 GM1 抗体,提示异质性免疫机制。
1.核心症状:
不对称性远端肌无力(90%):上肢首发占 80%,典型表现为单侧腕下垂、手指伸肌无力(“冷瘫”:寒冷诱发加重,见于 83% 患者)。
肌束颤动与痉挛(60%-80%):早期易误诊为 ALS,但无舌肌萎缩及锥体束征。
“肌无力先于萎缩”:病程 5 年内肌萎缩仅 30%,后期可出现 “爪形手” 等畸形。
2. 不典型表现:5% 患者近端肌无力,22% 诉下肢振动觉减退(无客观感觉神经传导异常),偶见臂丛 MRI T2 高信号(提示神经水肿)。
3. 伴随疾病:与自身免疫病(如强直性脊柱炎、克罗恩病)相关,HLA-DRB1*15 等位基因携带率增加。
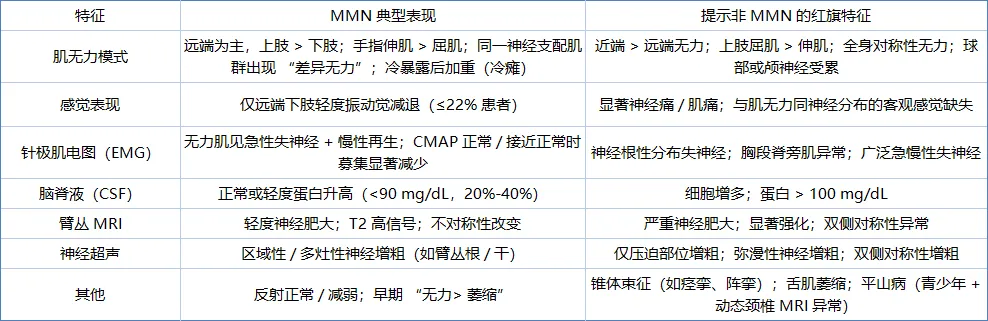
表 1:多灶性运动神经病(MMN)典型表现与提示其他诊断的 “红旗” 特征
多灶性运动神经病的诊断标准
1.确诊核心:
临床:≥2 个神经支配区不对称无力(持续>6 月),无客观感觉障碍。
电生理:2 条及以上运动神经存在明确传导阻滞(CMAP 面积↓≥50%,时限↑≤30%,远端 CMAP≥1mV),感觉传导正常。
支持证据:GM1 抗体阳性(滴度>1:1000)、IVIG 治疗有效、超声示神经局灶增粗(如正中神经前臂段直径>4.5mm)。
排除标准:上运动神经元体征、显著感觉障碍、球部受累、对称性全身无力。
2. 鉴别要点:
vs 运动神经元病(如 ALS):无锥体束征,神经超声无广泛神经萎缩,血清神经丝轻链正常。
vs 慢性炎症性脱髓鞘性多发性神经病(CIDP):CIDP 对称性、感觉运动均受累,传导阻滞伴广泛脱髓鞘特征;CSF 蛋白显著升高,脑脊液蛋白>1g/L;对激素敏感。
vs 平山病:青少年屈颈位 MRI 示硬膜囊前移,无传导阻滞。
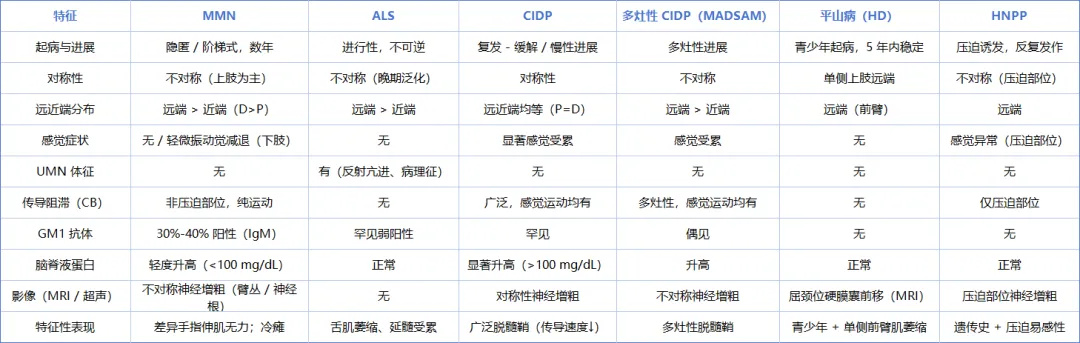
表 2:多灶性运动神经病(MMN)的鉴别诊断
多灶性运动神经病的治疗进展
1.一线方案:
IVIG:2g/kg 分 5 天,70%-94% 患者 4 周内起效,维持治疗(0.4g/kg 每 3-4 周)可延缓轴索丢失。预测疗效因素:年轻、CMAP 振幅>1mV、MRI 显示可逆性神经水肿。
SCIG:皮下注射(1-2g/kg 每周),疗效等同 IVIG,输注反应减少 50%,适合长期居家治疗。
2.二线策略:
利妥昔单抗:抗 CD20 单抗,对 GM1 抗体阳性者有效(ORR 65%),需监测 B 细胞重建。
补体抑制剂:依库珠单抗(抗 C5)Ⅱ 期试验显示,可减少传导阻滞区补体沉积,改善肌力。
3.禁忌与误区:激素 / 血浆置换可能加重病情(证据等级 1B),仅用于难治性病例需严格评估。
多灶性运动神经病的预后与监测
1.长期病程:10 年随访显示,50% 患者需轮椅辅助,早期治疗组轴索丢失速度降低 60%。
2.监测指标:每 6 个月复查神经传导(重点关注 CMAP 振幅)、肌肉超声(评估萎缩进展),GM1 抗体滴度与病情活动弱相关,不作为主要监测指标。
3.康复介入:早期物理治疗(如神经松动术)可预防挛缩,冷疗缓解 “冷瘫”,职业训练改善手功能(如使用加粗笔、防滑餐具)。
多灶性运动神经病的争议与前沿
1.病理争议:部分尸检发现前角细胞丢失,提示 “神经元 - 轴突 - 髓鞘” 三重损害,需重新审视 “纯周围神经病变” 理论。
2.新型靶点:抗 LINGO-1 抗体(促进髓鞘再生)、RNA 干扰技术(沉默补体 C3)进入临床前研究,未来或实现神经修复。
参考文献:
[1] Vlam L, Cats EA, Harschnitz O, et al. Complement Activity Is Associated With Disease Severity in Multifocal Motor Neuropathy. Neurology: Neuroimmunology and NeuroInflammation. 2023;10 (4):e119.
[2] Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on Management of Multifocal Motor Neuropathy. Journal of the Peripheral Nervous System. 2010;15 (4):295-301.
[3] Kleinveld VEA, Keritam O, Horlings CGC, et al. Multifocal Motor Neuropathy as a Mimic of Amyotrophic Lateral Sclerosis: Serum Neurofilament Light Chain as a Reliable Diagnostic Biomarker. Muscle and Nerve. 2024;69 (4):422-427.
[4] Yeh WZ, Dyck PJ, van den Berg LH, Kiernan MC, Taylor BV. Multifocal Motor Neuropathy: Controversies and Priorities. Journal of Neurology, Neurosurgery and Psychiatry. 2020;91 (2):140-148.
[5] Fitzpatrick AM, Mann CA, Barry S, et al. An Open Label Clinical Trial of Complement Inhibition in Multifocal Motor Neuropathy. Journal of the Peripheral Nervous System. 2011;16 (2):84-91.
[6] Herraets I, van Rosmalen M, Bos J, et al. Clinical Outcomes in Multifocal Motor Neuropathy: A Combined Cross-Sectional and Follow-Up Study. Neurology. 2020;95 (14):e1979-e1987.
[7] Bos JW, Groen EJN, Otten HG, et al. A 21-Bp Deletion in the Complement Regulator CD55 Promotor Region Is Associated With Multifocal Motor Neuropathy and Its Disease Course. Journal of the Peripheral Nervous System. 2024;29 (2):193-201.