髓鞘少突胶质细胞糖蛋白抗体相关疾病(MOGAD)的中国指南已于近日更新,本次更新是继2020年指南发布后在该领域的又一突破。让我们一起来看看本次指南更新了哪些重点吧!
髓鞘少突胶质细胞糖蛋白抗体相关疾病(MOGAD)是一种由靶向髓鞘少突胶质细胞糖蛋白(MOG)的免疫球蛋白 G(IgG)抗体介导的中枢神经系统炎性脱髓鞘疾病。其在病理学、影像学、治疗反应及结局等方面与多发性硬化(MS)、水通道蛋白 4 抗体阳性的视神经脊髓炎谱系疾病(AQP4+NMOSD)存在显著差异,是独立的疾病单元。本次指南更新从疾病基础与流行病学特征、临床特征与影像学表现、实验室诊断与鉴别诊断及后续治疗四个方面进行了更新。
MOG 是表达于少突胶质细胞膜的糖蛋白,约占中枢神经系统(CNS)髓鞘蛋白的 0.05%,富集于髓鞘板层外层。2007 年研究发现,识别 MOG 空间构象的抗体与非 MS 脱髓鞘疾病相关,目前以人全长MOG 基因转染细胞为基质的间接免疫荧光法或流式细胞术为公认的 MOG 抗体检测手段,且绝大多数 MOG 抗体为 IgG1 型。
MOGAD 的流行病学数据尚不完善,欧洲地区报道的发病率为(1.6~3.4)/100 万人。与 NMOSD(女性多发)和 MS(女性略多)不同,MOGAD 的男女发病比例无明显差异,可发生于各年龄阶段,<18 岁儿童和 20~30 岁是两个发病高峰。其临床表型具有年龄依赖性:<9 岁儿童多表现为急性播散性脑脊髓炎(ADEM),年长儿童(9~18 岁)及成人则以视神经炎、脊髓炎为主,且成人复发风险更高。约 50%~60% 的患者为复发型,反复发作可导致神经功能残疾累积。
MOGAD 的临床表型多样,主要包括视神经炎、脊髓炎及脑部综合征,不同类型可单独或组合发作,并反复发作。
(一)神经视炎
MOG 相关视神经炎(MOG - 视神经炎)是成人最常见的首发表现,可导致急性视力下降、严重视野缺损及色觉减退,常伴眼眶疼痛(眼球转动时加重)。体格检查可见相对性传入性瞳孔障碍(RAPD),急性期 45%~95% 的患者存在视盘水肿,严重者可出现视盘边缘放射状出血。起病时双侧同时受累者占 31%~58%,但复发时多为单侧。
影像学上,视神经 MRI 可见纵向延伸的长节段异常信号,好累及眶内段,伴视神经鞘膜及周围脂肪组织强化(“视神经周围强化”),部分患者可见眼球后壁强化。光学相干断层扫描(OCT)显示,急性期视盘周围视网膜神经纤维层(RNFL)显著增厚,数周后黄斑区神经节细胞层(GCL)先出现萎缩,多次发作后 RNFL 和 GCL 可明显萎缩,但视力多能维持一定水平,预后优于 AQP4 相关视神经炎。
(二)脊髓炎
MOG 相关脊髓炎表现为急性运动、感觉及括约肌功能障碍,尿潴留常早期出现,部分患者需保留导尿。≥50% 的患者急性期存在中度至重度运动障碍(扩展残疾状态量表评分 > 4 分),运动功能多可恢复,但可能遗留膀胱、肠道或性功能障碍。与 AQP4+NMOSD 不同,痛性痉挛、严重神经病理性疼痛少见。
MRI 上,脊髓病灶多纵向延伸≥3 个椎体节段,好发于下位胸髓、腰髓及圆锥,横断面累及中央灰质时呈 “H” 征,矢状位呈 “腹侧矢状线” 征,急性期可强化,伴脊膜或神经根强化。10% 的患者临床表现典型但 MRI 无可见病灶,且发作间歇期无症状脊髓病灶罕见(与 MS 不同)。
(三)脑部综合症
1、ADEM:是儿童 MOGAD 的典型首发表现,约 50% ADEM 患儿可检测到 MOG-IgG,成人仅占 5.6%。发病前常有呼吸道感染及发热,表现为头痛、局灶性神经功能缺损、脑病(意识下降、癫痫等),严重者需重症监护。MRI 可见多灶性、边界不清的 T₂高信号,伴中线结构或深部灰质受累,水肿可呈指状伸入皮质下,可见沿静脉走行的条片样强化及软脑膜强化。70% 以上 MOG抗体相关 ADEM 患者的临床症状和 MRI 病灶会完全或几乎完全消失。极少数患者MRI上脑白质病灶持续存在并逐渐融合,伴随逐渐加重的脑萎缩,临床表现为难治性癫痫和认知功能减退,结局较差。与 MS 不同,MOGAD 在发作间歇期出现无症状的脑部新病灶较少见,不需要通过系列MRI扫描监测MOGAD疾病活动。
2、脑干脑炎:多伴脑部多发病灶,40% 为无症状受累,有症状者表现为共济失调、复视。MRI 示中脑、脑桥或延髓弥漫性 T₂高信号,单侧或双侧小脑中脚病变具特征性,极后区受累及相关症状(恶心、呕吐等)较 AQP4+NMOSD 少见且轻微。
3、脑炎与皮质脑炎:少见但独特,约 6.7% 的 MOGAD 患者可出现,表现为癫痫、发热、意识减退等。MRI 液体衰减反转恢复(FLAIR)序列显示弥漫连续的皮质及皮质下异常信号,部分呈单侧 FLAIR 高信号伴癫痫(FLAMES 综合征),可伴软脑膜强化。
4、其他:MOGAD 可表现为孤立的或多发的颅内大病灶(假瘤样脱髓鞘),多发者在临床表型上和ADEM 有重叠。孤立的小脑半球发作少见,多为脑干病灶延伸而来,典型的累及一侧或双侧小脑中脚。极少数患者出现 MRI上持续存在的脑白质病变,伴进行性脑萎缩、难治性癫痫和认知功能下降。极罕见的情况下 ,MOGAD可有视神经以外的颅神经受累和脊神经根受累。
实验室诊断与鉴别诊断
(一)实验室检查
1、脑脊液检查:急性期呈炎性改变,>50% 患者细胞数增多(30%>50 个 /mm³),轻度至中度蛋白升高,15%~20% 可见寡克隆区带(OCB),但阳性率低于 MS 且多为一过性。
2、MOG-IgG 检测:
①推荐使用CBA,优先采用基于活细胞的 CBA(live-CBA),无法实施时可选固定细胞 CBA(fixed-CBA),不推荐酶联免疫吸附法(ELISA)。
②live-CBA与fixed-CBA滴度标准一致:滴度≥1∶100 为明确阳性,≥1∶10 且 < 1∶100 为弱阳性,高滴度诊断特异性更高。
③优先检测血清,高度怀疑者血清阴性时可查脑脊液。
④建议免疫治疗前检测,治疗后阴性者需 3 个月后或复发时复查。
高滴度 MOG-IgG 与MOGAD的临床特征密切相关,可将MOGAD与AQP4+ 视神经炎或 MS 区分开来。低滴度 MOG-IgG 也可见于MS、其他神经系统疾病和健康个体,需要仔细鉴别。需注意不同实验室检测的滴度不具有完全可比性,临床解释时需要谨慎。
3、合并其他神经系统自身抗体
①MOG-IgG 和 AQP4-IgG 同时阳性的情况非常罕见,必要时许更换实验室检测。
②如果在视神经炎、脊髓炎或其他符合 NMOSD 临床特征的患者血清中检出 AQP4-IgG,则 不需要再检测MOG-IgG;但 AQP4-IgG 阴性的患者推荐检测血清MOG-IgG。
③脑炎发作的 MOGAD 患者,可同时有脑脊液或血清抗 NMDAR 抗体阳性。
4、合并其他系统性自身抗体或自身免疫病
MOGAD较少合并其他器官特异性或非器官特性自身抗体及自身免疫病。用 CBA 检测法检出 MOG-IgG 阳性,排除其他可能的诊断,可确立MOGAD的诊断。

(二)诊断标准
需同时满足:
1、核心临床脱髓鞘事件(视神经炎、脊髓炎、ADEM 等);
2、MOG-IgG 阳性(明确阳性无需附加条件,弱阳性需 AQP4-IgG 阴性且满足 11 条影像附加条件之一。详见下图“推荐的MOGAD诊断标准”);
3、排除其他疾病。
(三)不支持诊断的“红旗征”
包括无发作时进行性神经功能恶化、急性发作时激素治疗无改善、MRI 符合 MS 特征(如边界清晰的 T₂病灶、OCB 持续阳性)等。

(四)诊断鉴别
1、与 AQP4+NMOSD:MOGAD 男女比例相当,儿童多见,脊髓圆锥易受累,极少合并其他自身免疫病;AQP4+NMOSD 女性占绝对优势,极后区综合征常见,合并自身免疫病多。
2、与 MS:MOGAD 发作间歇期无症状病灶罕见,OCB 阳性率 <20%;MS 多为复发 - 缓解型,OCB 阳性率> 75%,随时间累积无症状病灶。
(一)急性期治疗
静脉甲泼尼龙(IVMP):为首选,成人 1000mg/d 静脉输注 3~5 天,随后逐渐减量,口服激素减量需≥6 个月;儿童剂量 20~30mg・kg⁻¹・d⁻¹,参考成人方案递减(证据级别 2b,推荐级别 B)。
静脉免疫球蛋白(IVIG):用于激素反应不佳的严重发作,剂量 0.4g・kg⁻¹・d⁻¹,连续 5 天(证据级别 2b,推荐级别 B)。
血浆置换:激素无效者可采用,5~7 次,单次剂量为血浆容量的 1.0~1.5 倍,避免 IVIG 后立即使用(证据级别 2b,推荐级别 B)。
(二)缓解期序贯治疗
启动指征:
1、首次发作严重且恢复不佳(扩展残疾状态量表评分 > 2 分,视力 < 0.6);
2、发作≥2 次(证据级别 2b,推荐级别 B)。
常用药物包括:
1、硫唑嘌呤:2~3mg・kg⁻¹・d⁻¹,需联合激素至起效(4~5 个月)(证据级别 4,推荐级别 C)。
吗替麦考酚酯(MMF):初始 500mg bid,2 周后增至 750~1000mg bid(证据级别 2b,推荐级别 B)。
2、利妥昔单抗:按体表面积 375mg/m² 每周 1 次 ×4 周,或 1000mg×2 次(间隔 2 周)(证据级别 2b,推荐级别 B)。
3、托珠单抗:8mg/kg 静脉输注,每 4 周 1 次(证据级别 2b,推荐级别 B)。
4、间歇性 IVIG:每 4 周 0.4~2.0g/kg,儿童常用(证据级别 2b,推荐级别 B)。
治疗持续时间无明确标准,通常建议持续治疗 5 年无复发后,个体化评估是否停药。
(三)对症支持治疗
神经病理性疼痛可用普瑞巴林、加巴喷丁;尿潴留用溴比斯地明;痉挛状态用巴氯芬等。
关联项目
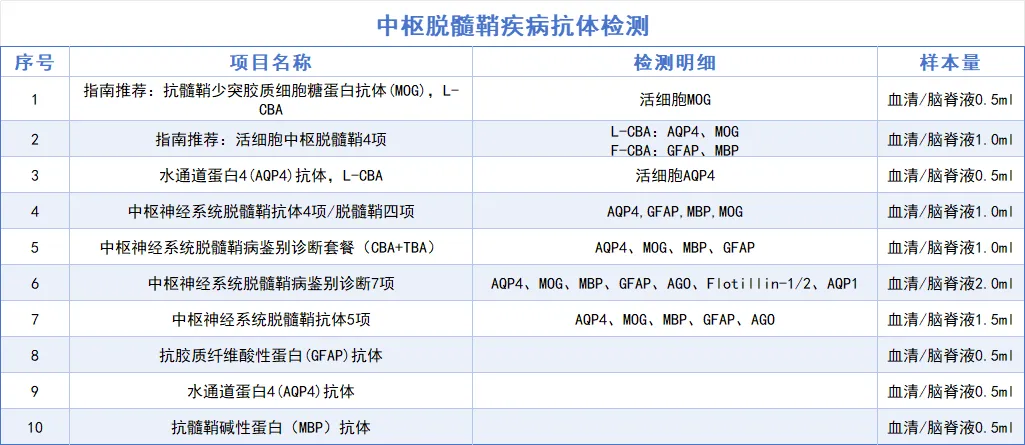
参考文献:中华医学会神经病学分会神经免疫学组. 中国髓鞘少突胶质细胞糖蛋白抗体相关疾病诊断与治疗指南(2025版)[J]. 中华神经科杂志,2025,58(07):704-720.